
Nature Protocols | 通用蛋白与核酸结构比对新算法US-align
蛋白质与核酸等生物大分子的三维结构比对是结构生物学和计算生物学中的核心任务之一,在功能注释、药物设计与进化研究中发挥着重要作用。近年来,深度学习结构预测模型和实验解析手段的迅猛发展,使高效、精准地对蛋白质、核酸及其复合物结构进行比对变得尤为关键。然而,现有大多数比对工具往往仅支持特定类型分子的比对,难以胜任跨分子、非序列顺序、循环排列等复杂结构的统一处理,限制了其在多样化研究场景中的应用。
近日,中国科学院深圳先进技术研究院张成辛研究员,密歇根大学Lydia Freddolino教授、新加坡国立大学张阳教授团队在Nature Protocols上发表研究论文“A graphic and command line protocol for quick and accurate comparisons of protein and nucleic acid structures with US-align”,系统介绍了他们开发的通用结构比对工具US-align(https://zhanggroup.org/US-align/)。该算法支持蛋白质、DNA、RNA及其复合物的快速统一比对,具备命令行、PyMOL插件和网页服务器三种使用方式。

文章上线截图
原文链接:https://doi.org/10.1038/s41596-025-01189-x
US-align算法构建于张阳团队早期提出的TM-score和TM-align基础上,后者已广泛用于蛋白质结构质量评估和结构比对任务。与之相比,US-align不仅支持蛋白,还扩展至核酸、蛋白–核酸复合物甚至小分子的比对,并于2022年以核心算法形式发表于Nature Methods。相较于2022年发表的纯命令行版本,新的论文在US-align功能实用性和用户体验上有显著扩展和增强:新增了循环排列结构比对模块,支持多结构比对任务(图1),增强了结果可视化能力。用户可通过PyMOL插件(图2)、网页版服务器(图3)或命令行执行比对任务,命令行结果支持在PyMOL、UCSF ChimeraX、RasMol等多种分子可视化软件中查看(图4)。此外,作为一款可同时完成序列顺序的结构比对与非序列顺序比对的算法,最新版本的US-align还创新性地开发一套全新的循环排列(Circular Permutation)结构比对算法(图5),是第一款基于TM-score打分函数的循环排列比对程序。此外,新版US-align还支持批量结构比对,为三维结构数据库搜索提供便利。
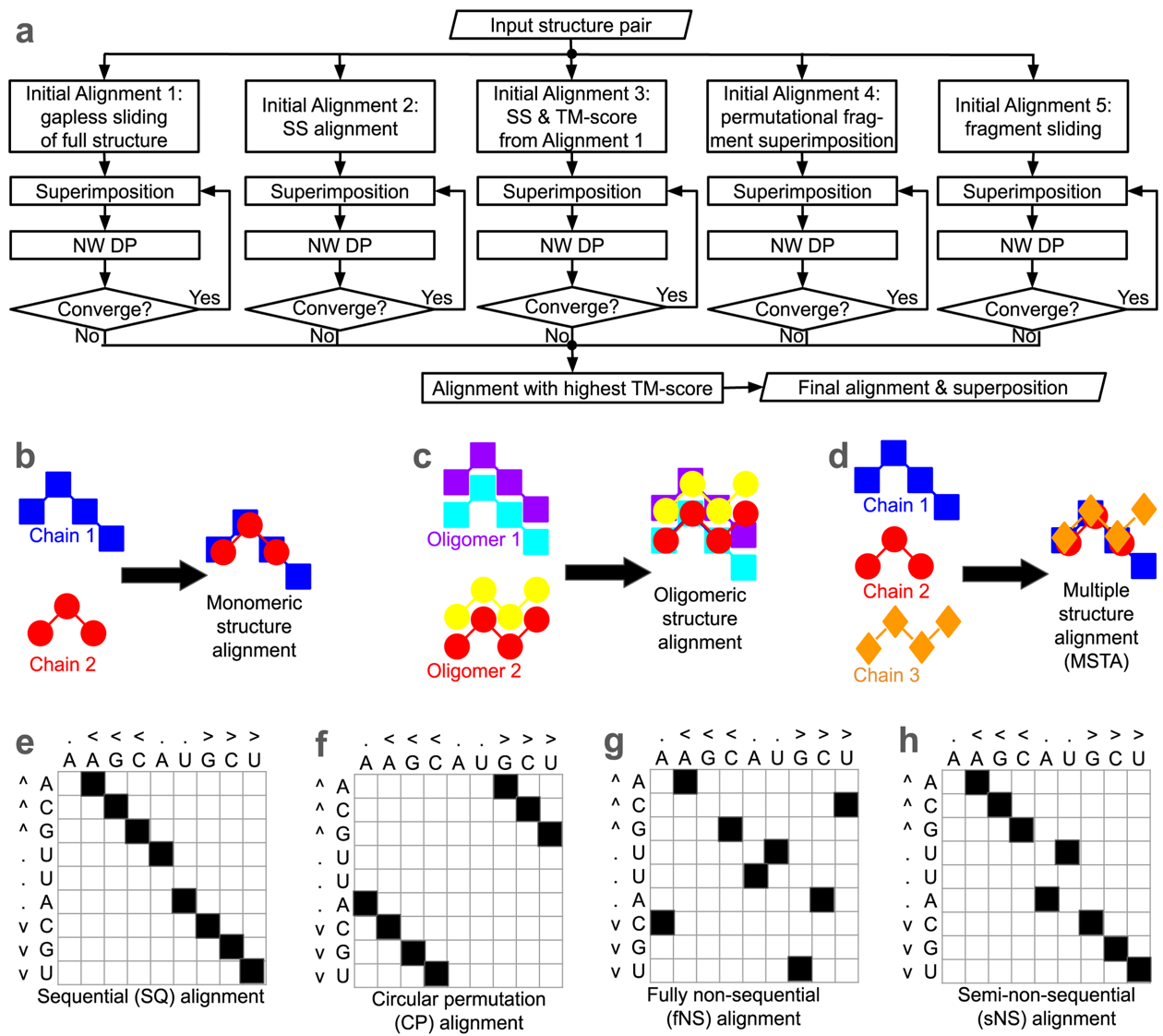
图1. US-align的算法流程图(a)与可执行的三维结构比对任务,包括b, 单体结构两两比对;c, 复合物结构两两比对;d, 多结构比对;e, 序列顺序比对;f, 循环顺序比对;g, 完全非序列顺序比对;h, 部分非序列顺序比对。
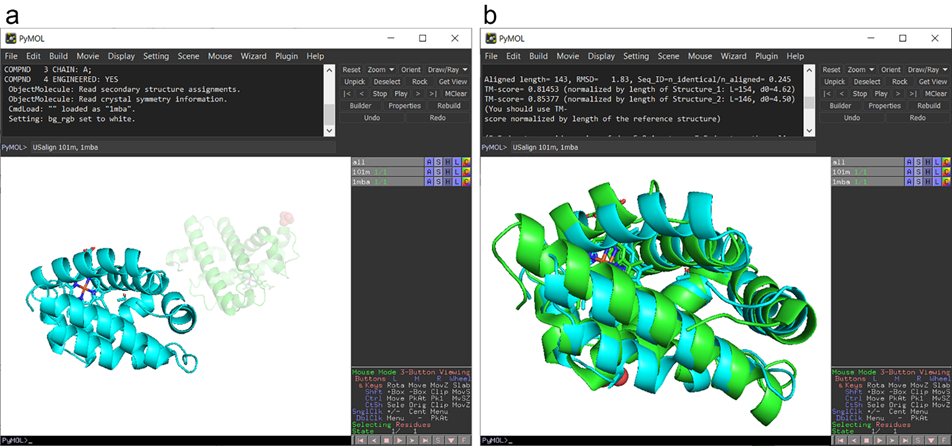
图2. 通过PyMOL插件执行US-align结构比对任务。a, 比对前。b,比对后。
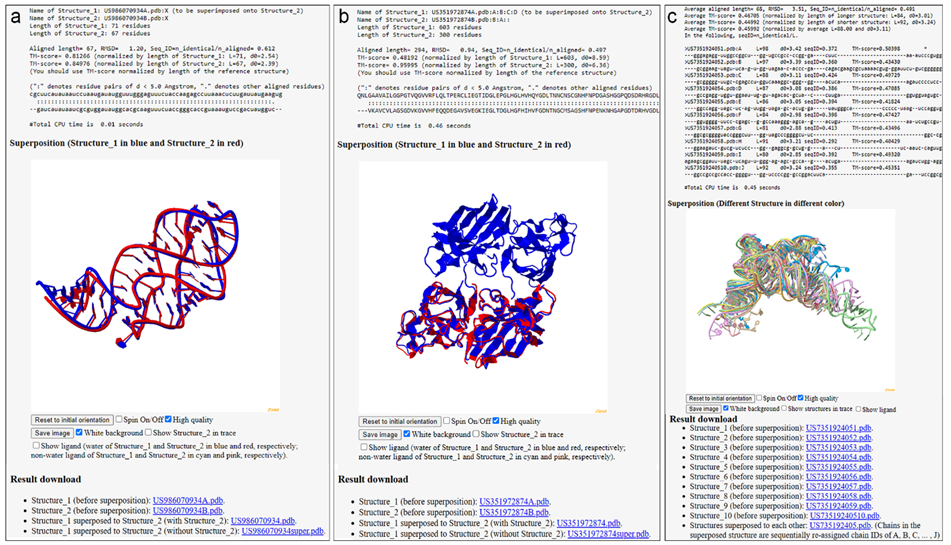
图3. 通过网页服务器执行US-align比对任务的网页输出。a, 单体RNA结构的两两比对。b, 蛋白复合物的两两比对。c, RNA的多结构比对。
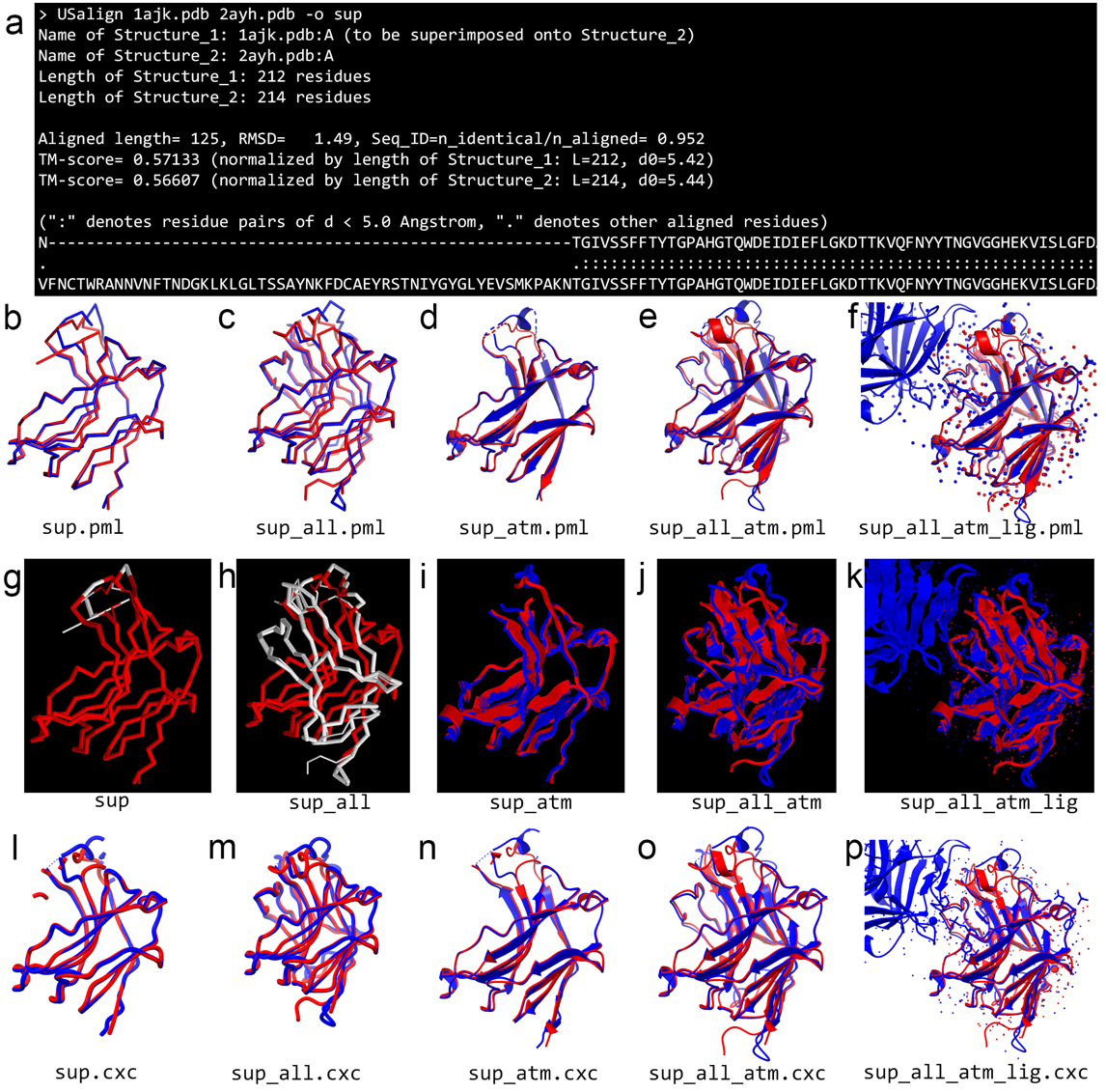
图4. US-align命令行程序两两比对结果(a)在PyMOL(b-f)、RasMol(g-k)与UCSF ChimeraX(l-p)分子可视化程序中不同风格的三维结构展示。
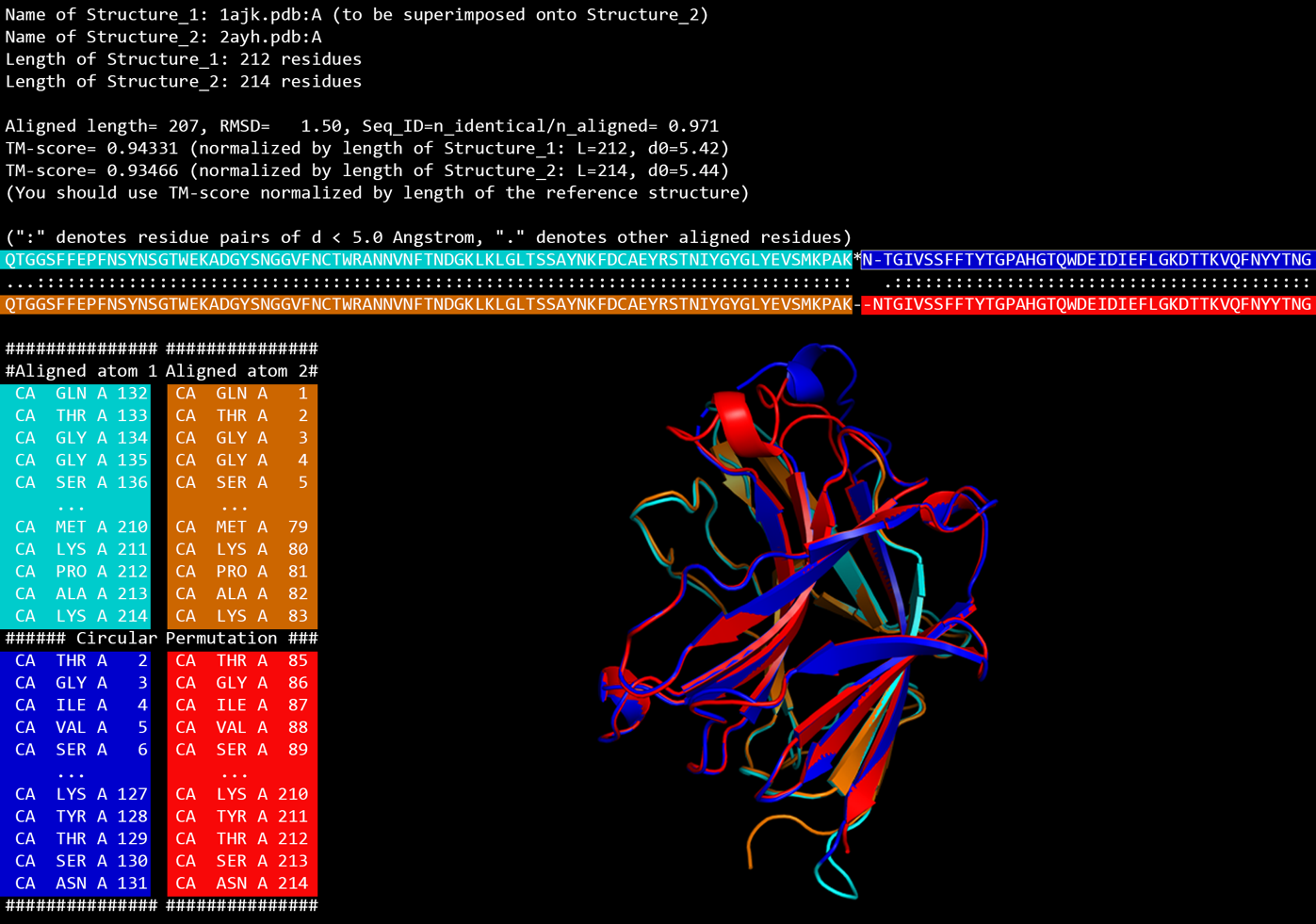
图5. US-align进行循环排列结构比对的实例。
未来展望
US-align 是目前少数可同时处理单体、复合物、多结构、序列和非序列顺序比对,以及循环排列比对任务的通用结构工具。它以统一的TM-score打分函数描绘蛋白与核酸结构间的相似性,并配套多种可视化和批量处理功能,为科研人员提供了高效、灵活、直观的结构分析平台。相较于2022年开发的早期纯命令行版本,最新版本的US-align加入了循环排列结构比对与批量结构比对模块,并且支持PyMOL、UCSF ChimeraX、RasMol以及网页服务器等多种可视化方式,使得复杂多样的三维结构比对任务得以快速、高效与直观地进行。目前,对于构象变化较大的蛋白与核酸结构,US-align尚未能进行有效的柔性结构比对,开发新型的柔性结构比对算法将是US-align平台的未来研发方向之一。
参考文献
1. Zhang C, Freddolino L, Zhang Y (2025). “A graphic and command line protocol for quick and accurate comparisons of protein and nucleic acid structures with US-align.” Nature Protocols, https://doi.org/10.1038/s41596-025-01189-x.
2. Zhang Y, Skolnick J (2004). “Scoring function for automated assessment of protein structure template quality.” Proteins, 57: 702-710.
3. Zhang Y, Skolnick J (2005). “TM-align: A protein structure alignment algorithm based on TM-score.” Nucleic Acids Research, 33: 2302-2309
4. Zhang C, Shine M, Pyle AM, Zhang Y (2022). “US-align: Universal Structure Alignments of Proteins, Nucleic Acids, and Macromolecular Complexes.” Nature Methods, 19: 1109–1115.
5. Zhang C, Pyle AM (2022). “A unified approach to sequential and non-sequential structure alignment of proteins, RNAs and DNAs.” iScience, 25: 105218.