

In existing technologies, experimental methods such as flow cytometry and single-cell RNA sequencing (scRNA-seq) can directly determine the proportion of cells in the tumor microenvironment, but these methods are usually costly. To solve this problem, researchers have developed various computational methods to estimate the proportion of different types of cells based on low-cost bulk RNA sequencing (bulk RNA-seq) data, such as EPIC, MuSiC, CIBERSORTx, Scaden, Kassandra, etc. However, due to the large differences in gene expression profiles of cancer cells in different tumor types, using a single model to accurately predict the abundance of various cells in multiple tumors remains a challenge.
In response to this problem, the team of Associate Researcher Li Xuefei from the Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences, and Associate Professor Tian Liang from Hong Kong Baptist University, have developed a deconvolution algorithm based on deep learning and publicly available single-cell datasets. This algorithm can accurately estimate the abundance of 16 cell types in 19 types of solid tumors. The related research results were published online on November 8, 2024, in the PNAS magazine with the title "DeSide: A unified deep learning approach for cellular deconvolution of tumor microenvironment".

Article Online Screenshot
Original Article Link: www.pnas.org/doi/10.1073/pnas.2407096121
Main Innovations
Synthesizing High-Quality Training Sets
Firstly, this study integrated 12 scRNA-seq datasets from 6 types of solid tumors to provide a more comprehensive reference for synthesizing virtual tumor bulk RNA-seq data (training set). Secondly, this study proposed a new sampling method, segment sampling, which makes the synthesized virtual tumor bulk RNA-seq data contain a more diverse combination of cell proportions. In addition, when synthesizing bulk RNA-seq data, this study retains genes highly related to each cell type through gene-level filtering to reduce the dimensionality of the input data; and retains samples with high similarity to real tumor expression profiles through GEP-level filtering. These innovations together effectively improve the quality of the synthesized virtual tumor bulk RNA-seq data.
Innovations in Deep Neural Network Structure
In the deep neural network (DNN) structure, DeSide innovatively uses two fully connected networks: the pathway network and the GEP network, which extract feature information from biological signal pathways and gene expression profiles (GEP), respectively. The pathway network introduces coarse-grained features, effectively enhancing the diversity of the input data.
Considering the large differences in gene expression profiles of tumor cells between different cancer types, DeSide uses the sigmoid function as the activation function of the DNN output layer, keeping the output sum within the [0,1] interval. This design first predicts the proportion of non-cancer cells such as immune cells, and then estimates the proportion of tumor cells by subtracting the proportions of all non-cancer cells from 1, effectively reducing the errors brought by directly predicting the proportion of tumor cells. It is worth noting that DeSide is the first algorithm to introduce this strategy in the direction of using deep neural networks to predict cell proportions.
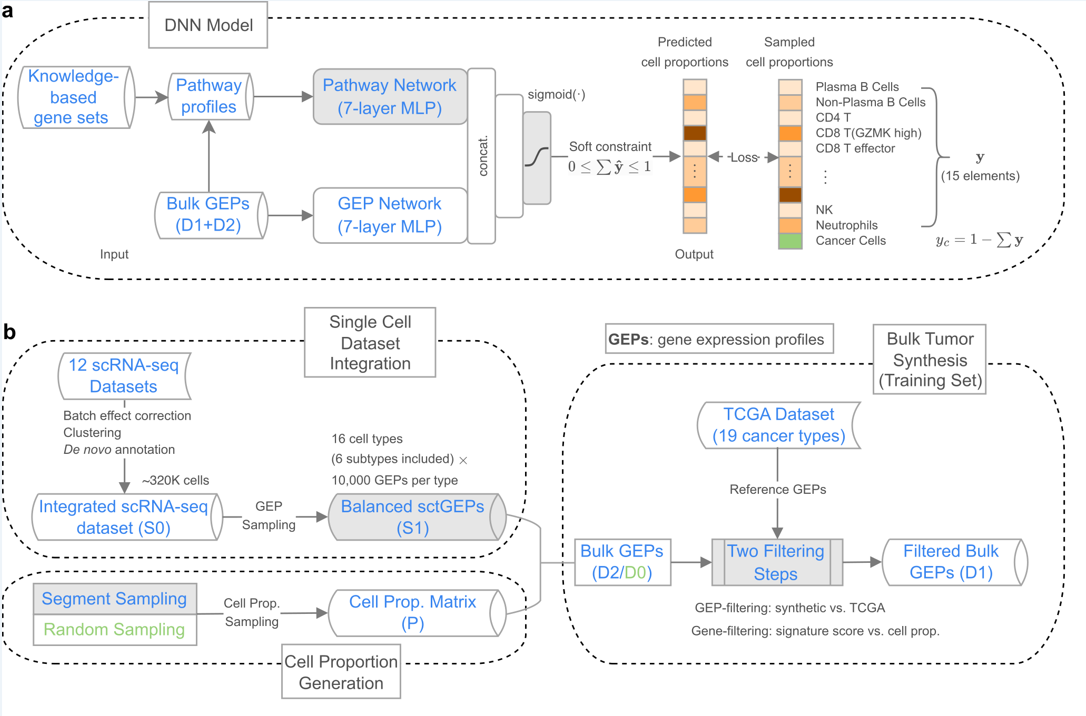
Figure 1. (a) The structure of the DeSide deep neural network (DNN) model; (b) The process of synthesizing the bulk RNA-seq gene expression profiles (GEP) of virtual tumors.
Comparison of Prediction Effects with Other Algorithms
The study systematically compared the accuracy of DeSide with existing algorithms in predicting the proportion of cells in the tumor microenvironment. The results show that DeSide can well predict the proportion of different cell types in various tumor types (Figure 2a, b). At the same time, even compared with reference-based models, DeSide's performance on specific tumor types is still excellent (Figure 2c). It is worth noting that DeSide can accurately predict cancer types not included in the training set, showing good generalization ability.
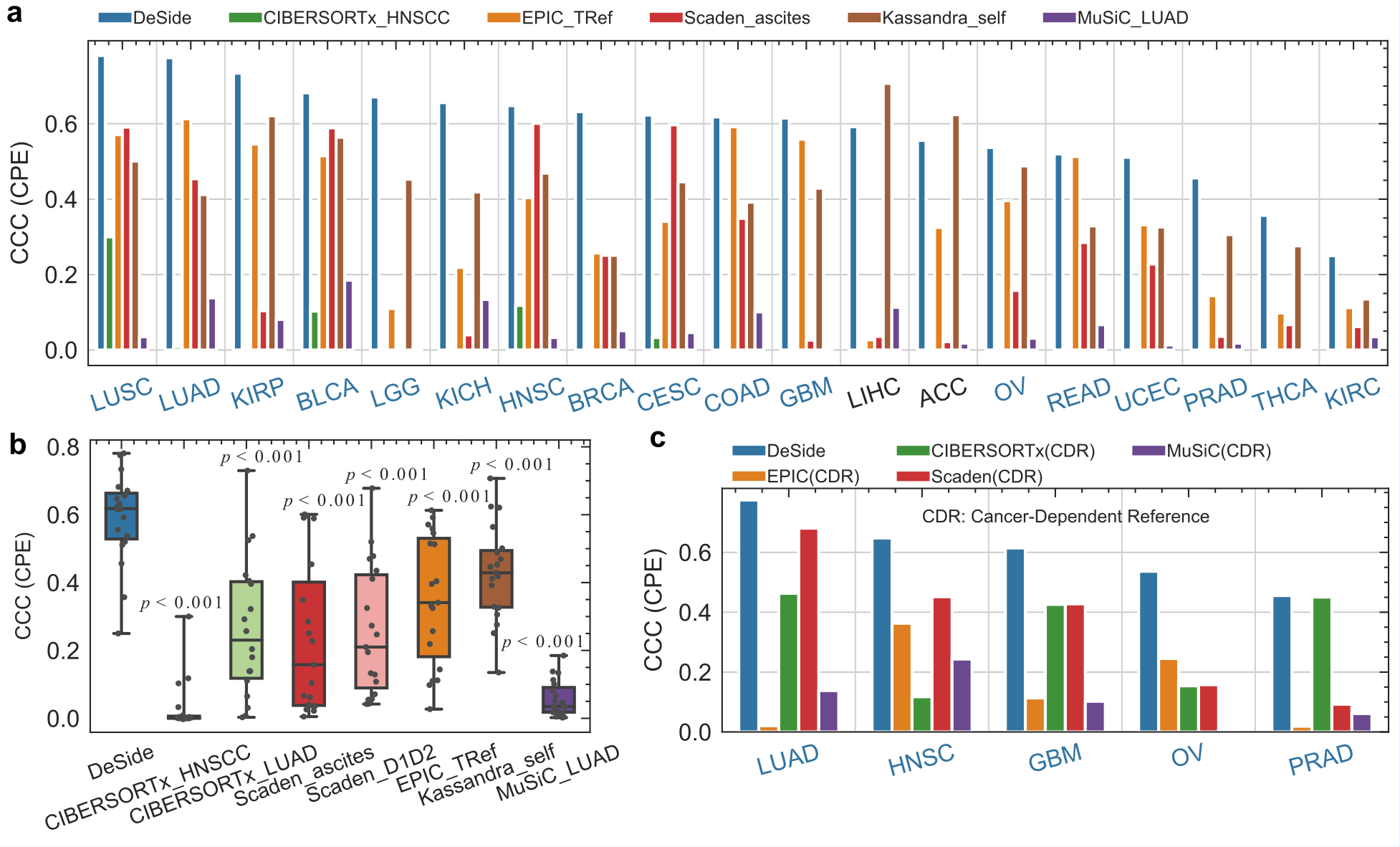
Figure 2. Comparing the ability of DeSide and other algorithms to predict the proportion of tumor cells in bulk RNA-seq data. CCC refers to the concordance correlation coefficient (CCC) between the predicted proportion of tumor cells and the tumor purity estimated based on gene copy numbers. Data is sourced from The Cancer Genome Atlas (TCGA) database.
The Value of DeSide in Clinical Prognosis Analysis
The study explored the potential value of DeSide in clinical applications through patient survival analysis. The results show that the cell proportions predicted by DeSide can effectively classify patients according to disease progression, that is, the abundance of certain cell types or their combinations is significantly related to patient survival (Figure 3). In the future, DeSide is expected to further help explore the key interactions between different cells, providing new possibilities for finding potential clinical treatment targets.
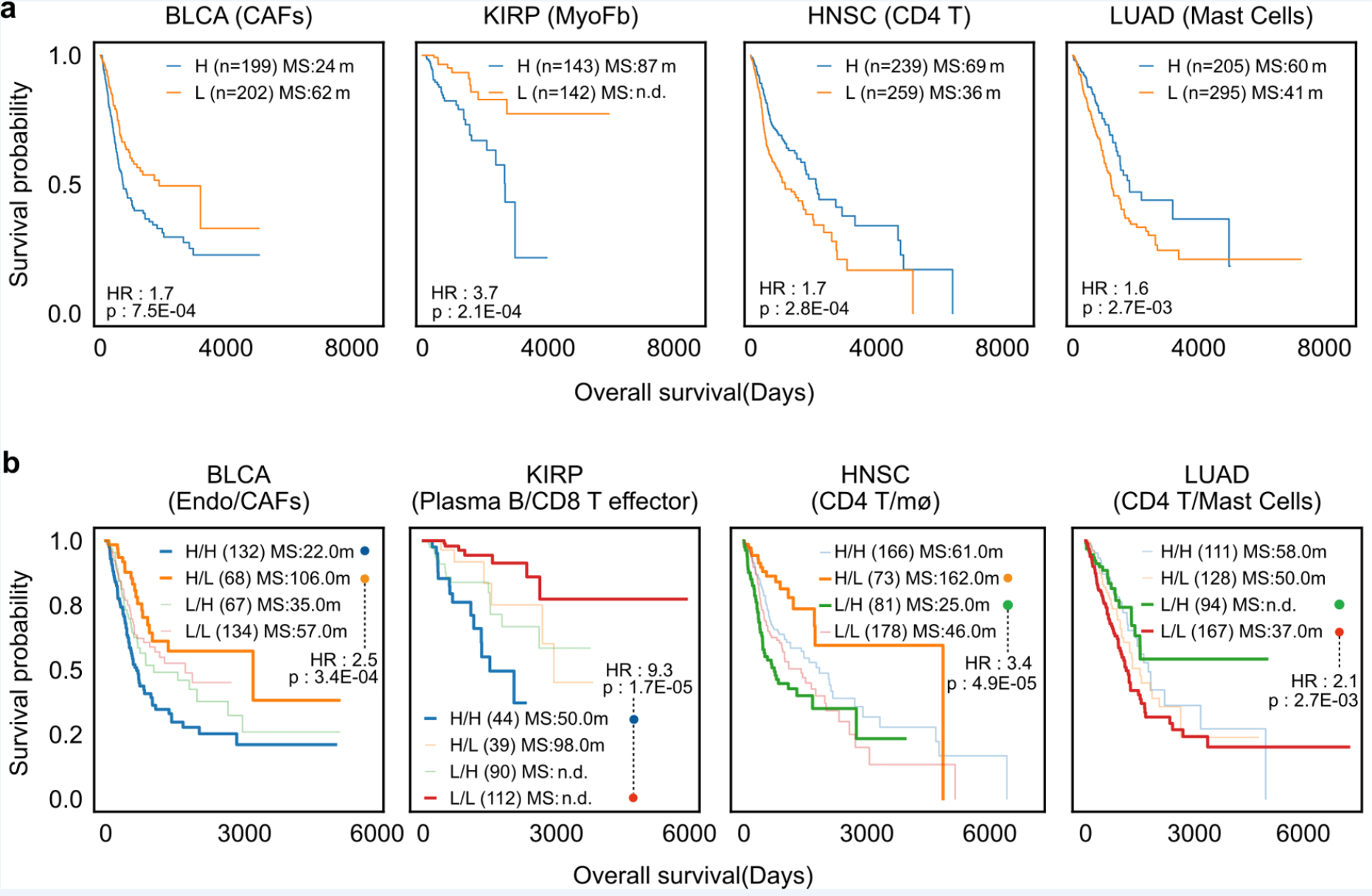
Figure 3. Based on DeSide's prediction of the proportions of different cell types within various tumors from public datasets, patients are classified and subjected to survival analysis.
Conclusion and Outlook
The DeSide algorithm developed in this study, based on publicly available single-cell datasets and deep learning methods, can accurately and efficiently estimate the proportions of 16 cell types in bulk RNA-seq samples of 19 types of solid tumors. This provides a powerful method and data support for deepening the understanding of tumor occurrence and development mechanisms, assessing patient prognosis, and formulating precise treatment strategies.
Associate Researcher Li Xuefei from the Synthetic Biology Evolution Research Center of the Synthetic Biology Institute, Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences, and Associate Professor Tian Liang from the Department of Physics, Hong Kong Baptist University, are the co-corresponding authors of this article. Fourth-year doctoral student Xiong Xin from Hong Kong Baptist University and Research Assistant Liu Yerong from the Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences, are the co-first authors of this article. Xiong Xin initiated the research during his tenure as a research assistant in Li Xuefei's research group in 2020-2021, and later, recommended by Li Xuefei, went to Tian Liang's research group to pursue a doctoral degree. The two research groups closely collaborated to complete this study. This work was supported by multiple projects including the National Key R&D Program, the Strategic Priority Research Program of the Chinese Academy of Sciences, the National Natural Science Foundation for Young and General Projects, the Guangdong Basic and Applied Basic Research Foundation, and the Hong Kong Research Grants Council, among others.